faculty lab
The Tibbetts Lab
The Tibbetts Lab studies growth control and DNA repair pathways that become disrupted in cancer cells while maintaining secondary interests in neurodegenerative disease.
DNA repair defects and increased proliferative potential are two hallmarks of virtually all cancer cells. We are investigating pathways controlling DNA repair and cell proliferation—and the interfaces between them—to define cancer cell vulnerabilities that may be exploited through novel targeted therapies.
Last updated:
Genome surveillance and cell growth regulation
There are currently two active projects that broadly pertain to mechanisms of genome surveillance growth regulation and tumor suppression.
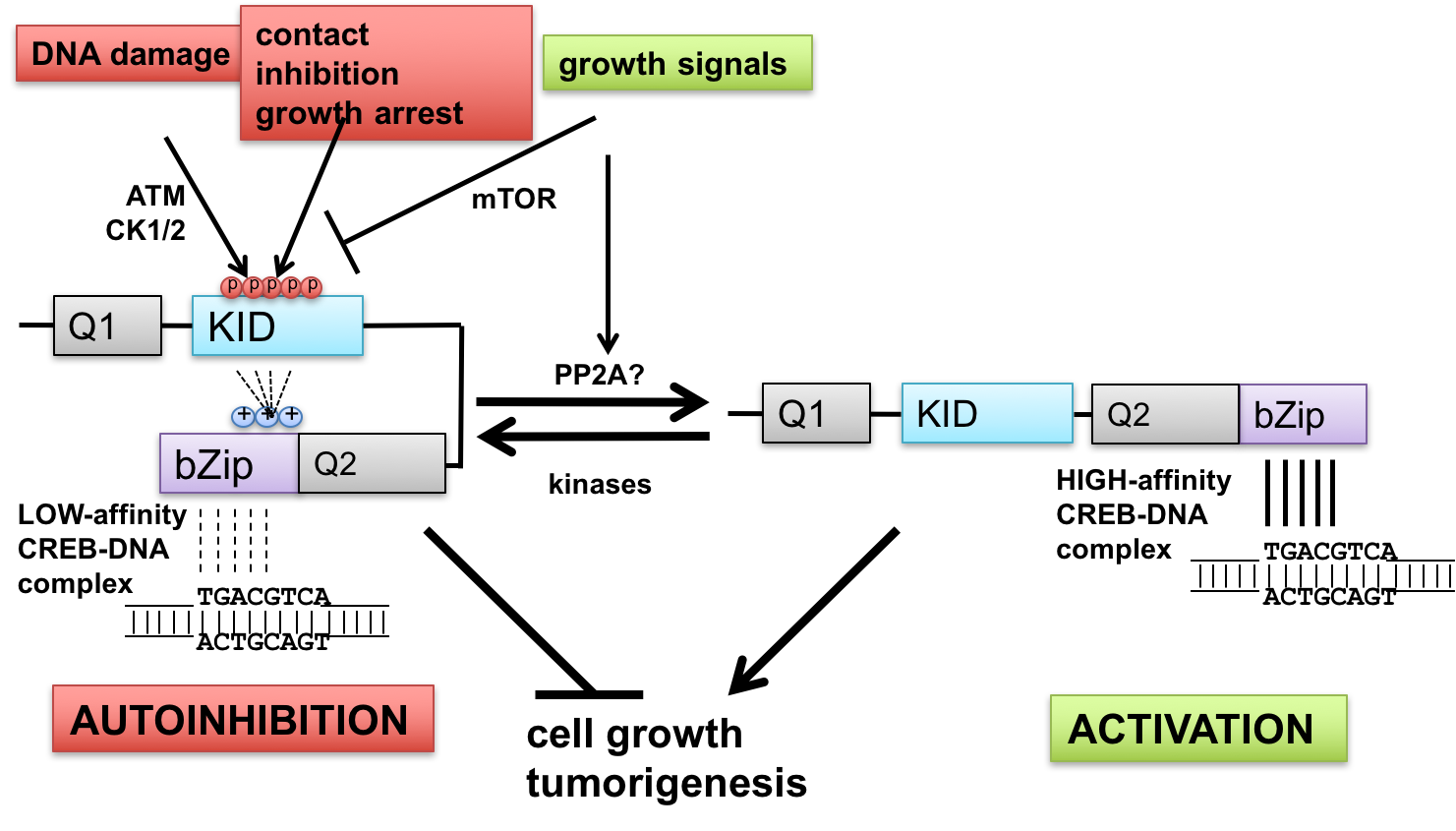
- Interface between DNA damage signaling and CREB-mediated transcription: implications for tumor suppression and metabolic control—The ataxia-telangiectasia-mutated (ATM) protein kinase and related proteins ATR and DNA-PK function as master regulators of the cellular DNA damage response, phosphorylating hundreds of proteins in response to various types of genotoxic stress. Mutations in ATM cause ataxia-telangiectasia (A-T), a syndrome of cancer susceptibility and neurodegeneration characterized at the cellular level by radiation sensitivity and dramatically impaired ability to signal and repair DNA double-strand breaks. A-T patients and ATM-deficient mice also manifest metabolic abnormalities suggesting that ATM senses and responds to metabolic cues. We are investigating how ATM signaling through the CREB (cAMP-response element-binding protein) contributes to cell growth and metabolic control. CREB fulfills key roles in metabolism (gluconeogenesis) and long-term memory formation and has been strongly implicated as a protooncogene in a host of cancers. We have defined a new mechanism of CREB regulation whereby ATM-dependent phosphorylation of a conserved cluster of Ser/Thr residues diminishes CREB DNA binding activity through an autoinhibitory mechanism. In addition, ATM-independent pathways mediate CREB autoinhibition in response to a variety of growth-inhibitory stimuli, whereas signaling through the pro-growth mTOR pathway promotes CREB dephosphorylation (Fig. 1). In addition to defining these pathways and elucidating biochemical mechanisms of autoinhibition, we are currently investigating functional impacts of cancer-associated mutations that may diminish CREB autoinhibition. The importance of these pathways in tumor suppression and other CREB-mediated processes is being tested using CREB gene-edited mice.
- Novel roles of RNA-binding proteins in DNA damage repair and tumor suppression—A second project is focusing on emergent roles for RNA-bindings proteins (RBPs) in DNA damage-dependent alternative splicing, DNA repair and tumor suppression. We are elucidating pathways controlling DNA damage-dependent changes in alternative splicing and modeling the implications of these changes in mice using CRISPR/CAS9-mediated genome editing in mice. The long-term goal of these studies is to understand fundamental aspects of DNA damage repair and response that can be used to guide radio- and chemotherapeutic strategies for cancer patients.
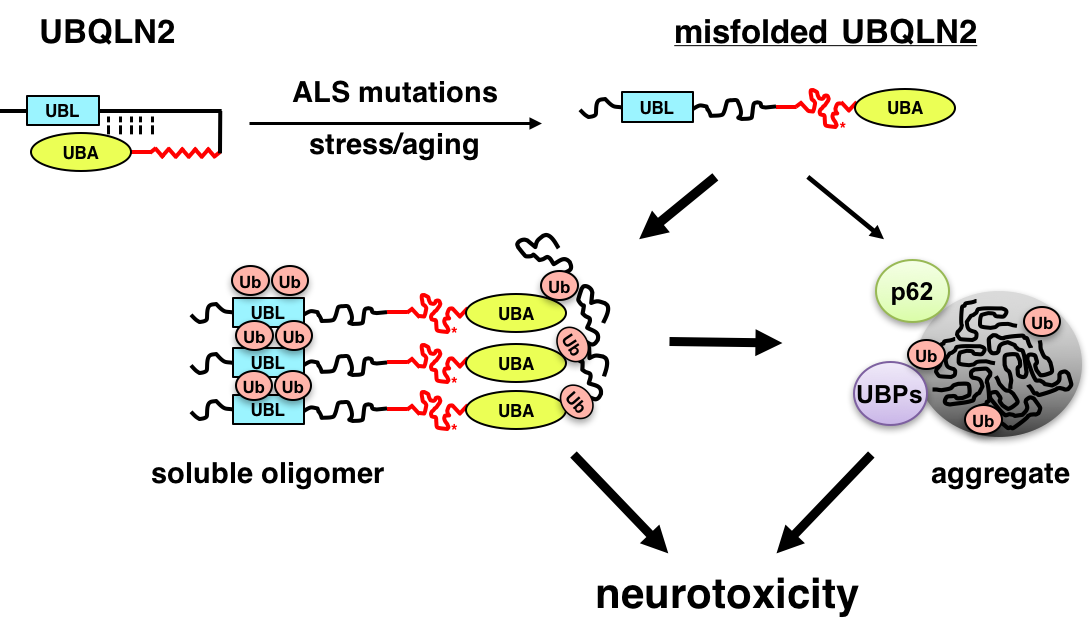
Molecular pathogenesis of ALS
ALS is a fatal neurodegenerative disease that affects motor neurons. Recent genetic advances have identified genes that are critically involved in the ALS disease process, including the RNA-binding proteins TDP-43 and FUS/TLS, the uncharacterized open reading frame, C9ORF72, and UBQLN2. We are using cell culture, Drosophila melanogaster (fruit fly) and mouse models to understand how ALS-associated mutations in these genes instigate neurodegeneration. We are particularly interested in how ALS mutations in the ubiquitin chaperone UBQLN2 promote its misfolding and aggregation, leading to disruption of protein clearance mechanisms and neuron death (Fig. 2). The long-term goal of this work is to identify key pathways that may be amenable to therapeutic intervention in ALS and related dementias.